Input data and parameters
QualiMap command line
| qualimap bamqc -bam /data/qualimap_release_data/alignments/HG00096.chrom20.bam -c -nw 400 -hm 3 |
Alignment
| BAM file: | /data/qualimap_release_data/alignments/HG00096.chrom20.bam |
| Program: | GATK IndelRealigner (1.0.4487) |
| Size of a homopolymer: | 3 |
| Number of windows: | 400 |
| Skip duplicated alignments: | no |
| Analysis date: | Wed Sep 02 09:28:03 CEST 2015 |
| Draw chromosome limits: | yes |
Summary
Globals
| Reference size | 63,025,520 |
| Number of reads | 2,939,810 |
| Mapped reads | 2,914,581 / 99.14% |
| Unmapped reads | 25,229 / 0.86% |
| Paired reads | 2,904,345 / 98.79% |
| Mapped reads, only first in pair | 1,453,937 / 49.46% |
| Mapped reads, only second in pair | 1,450,408 / 49.34% |
| Mapped reads, both in pair | 2,896,042 / 98.51% |
| Mapped reads, singletons | 8,303 / 0.28% |
| Read min/max/mean length | 100 / 100 / 100 |
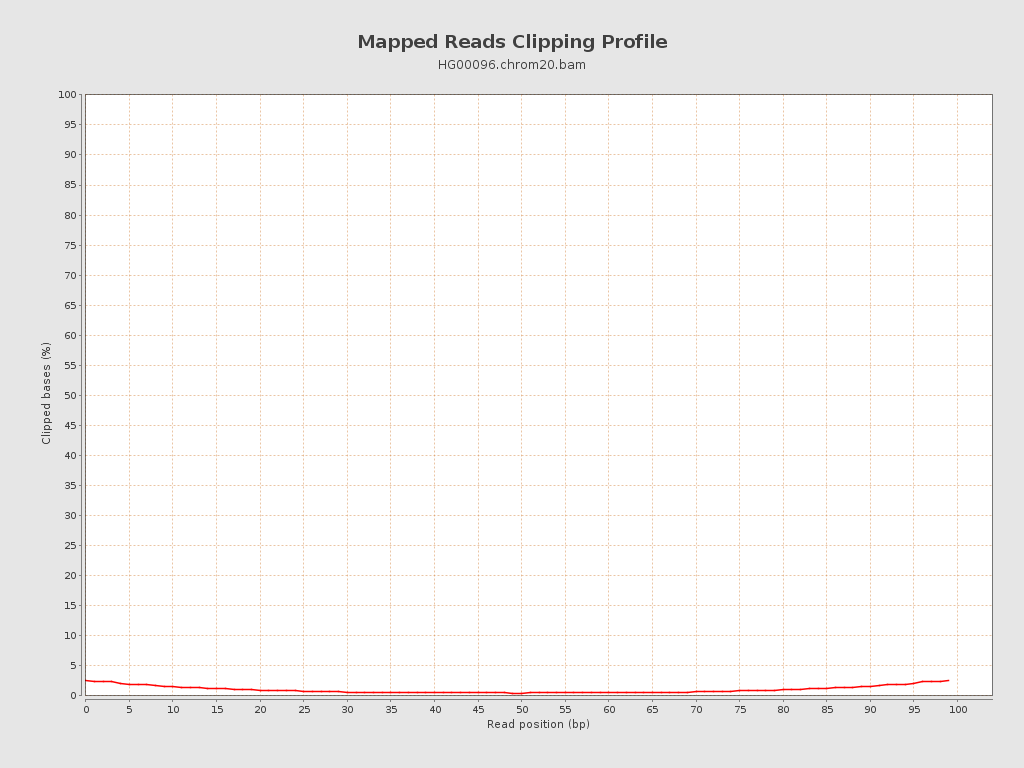
| Clipped reads | 678,220 / 23.07% |
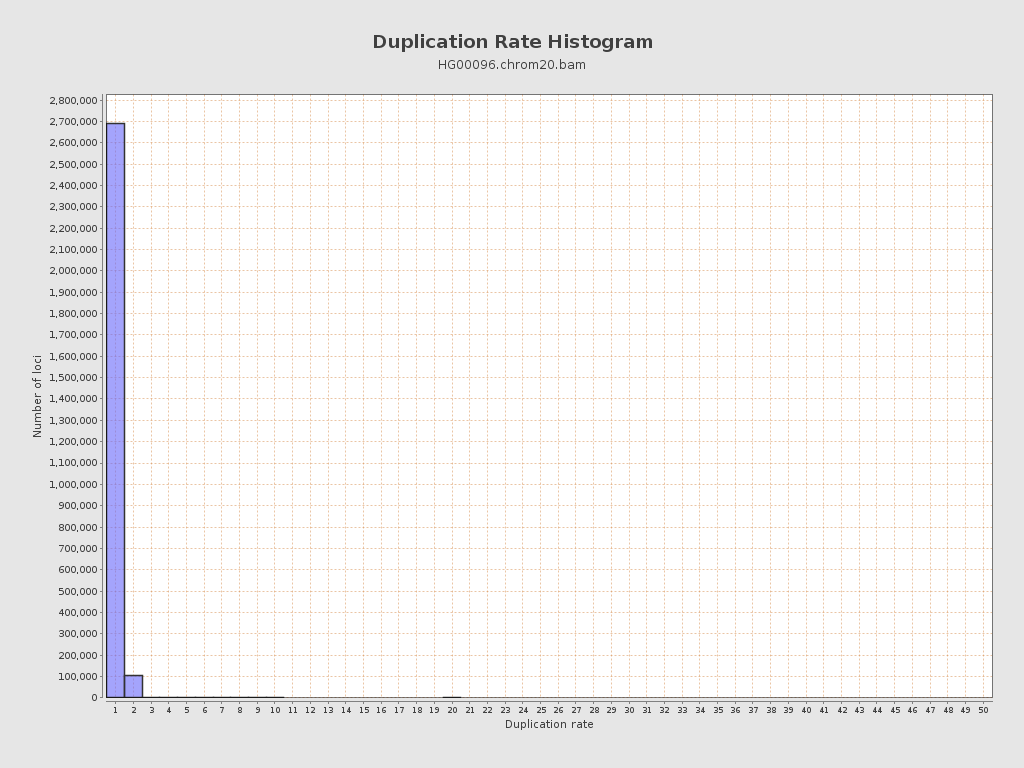
| Duplicated reads (flagged) | 39,111 / 1.33% |
ACGT Content
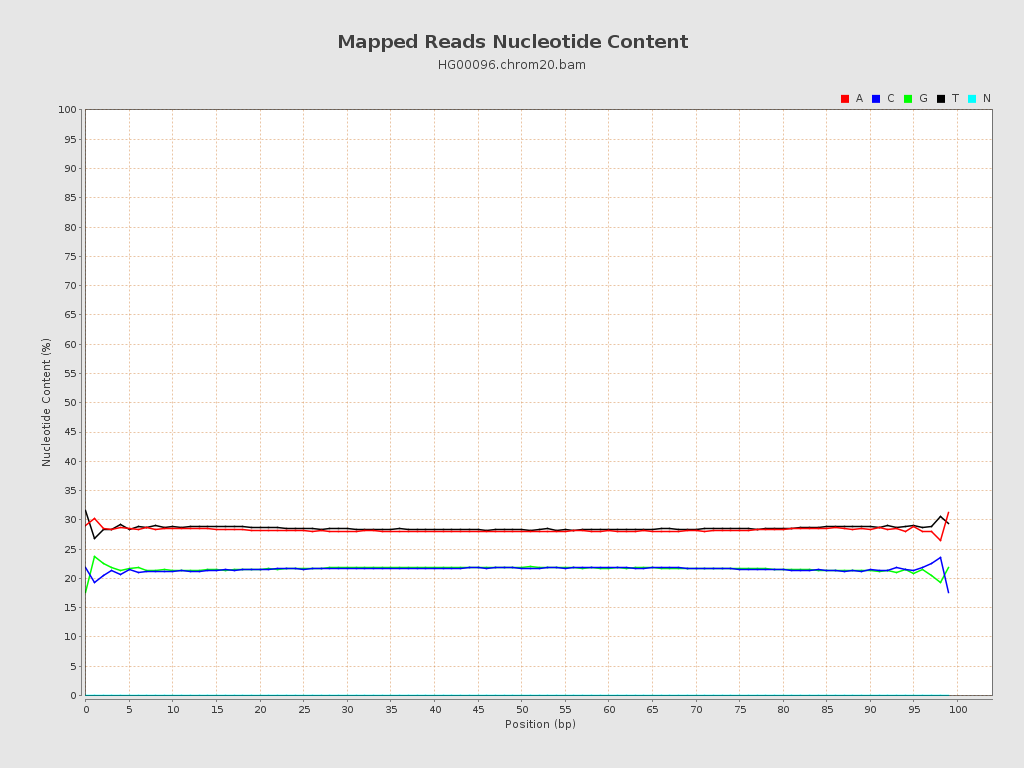
| Number/percentage of A's | 78,482,215 / 28.24% |
| Number/percentage of C's | 59,888,941 / 21.55% |
| Number/percentage of T's | 79,422,510 / 28.58% |
| Number/percentage of G's | 60,086,521 / 21.62% |
| Number/percentage of N's | 0 / 0% |
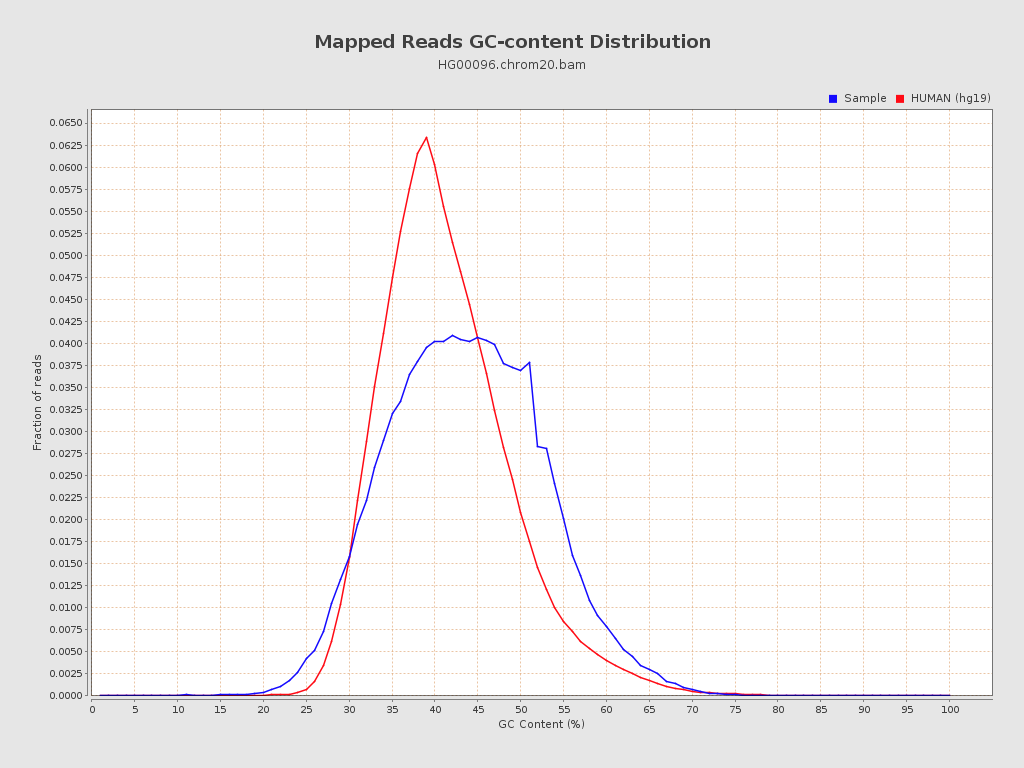
| GC Percentage | 43.18% |
Coverage
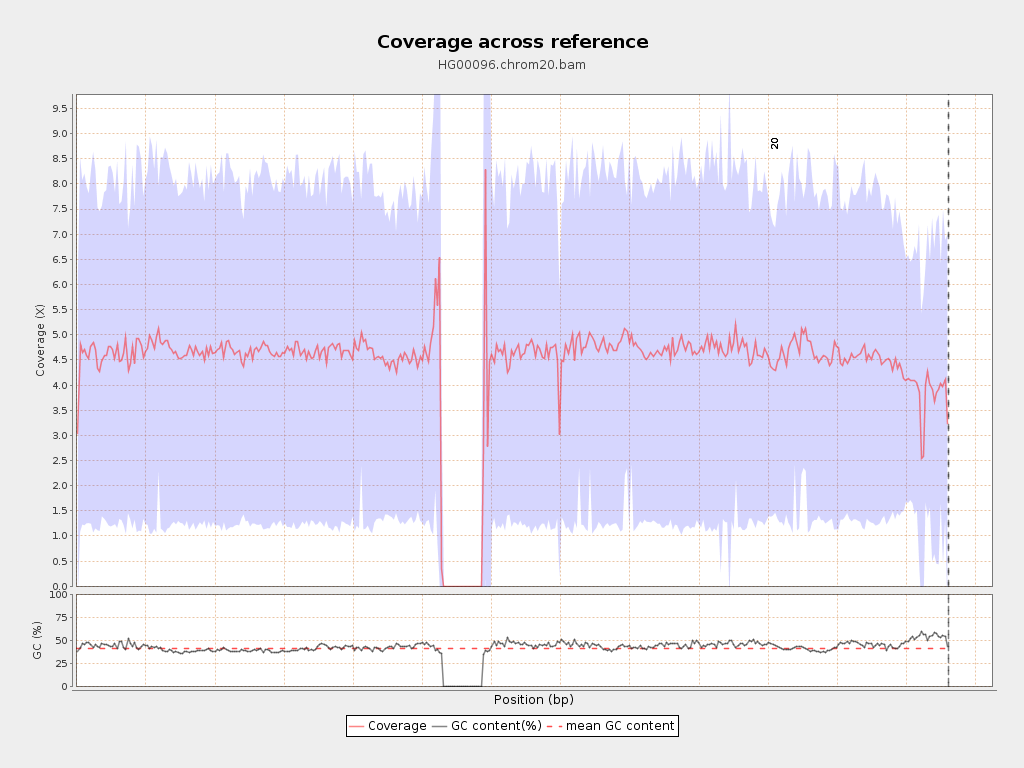
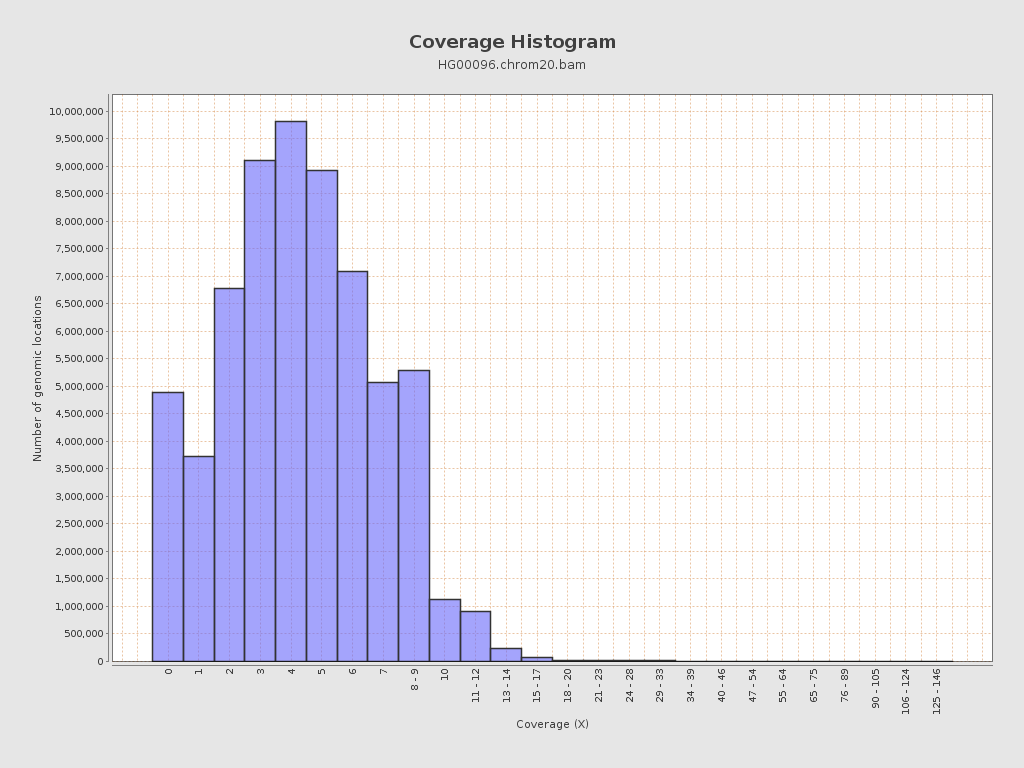
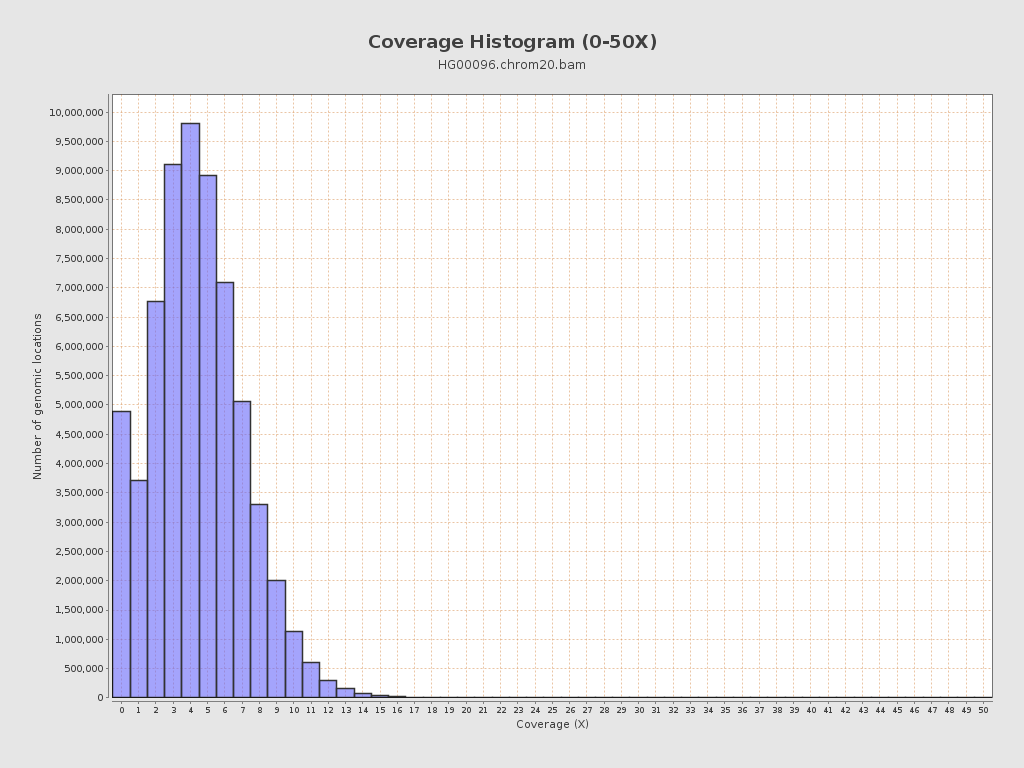
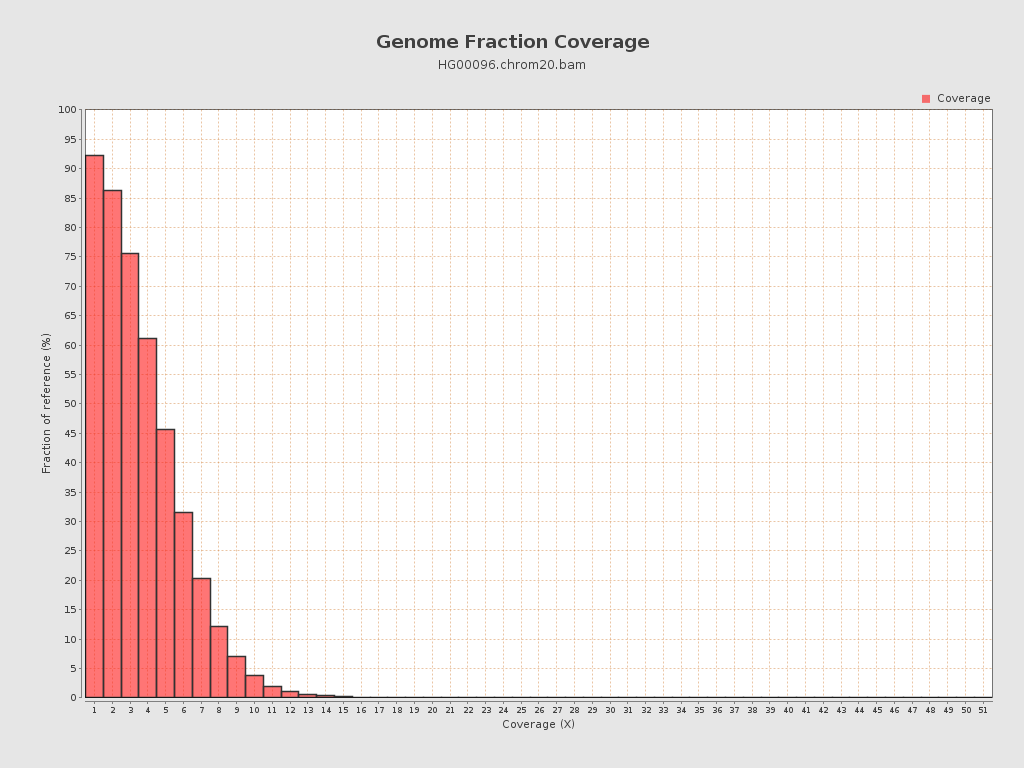
| Mean | 4.41 |
| Standard Deviation | 2.75 |
Mapping Quality
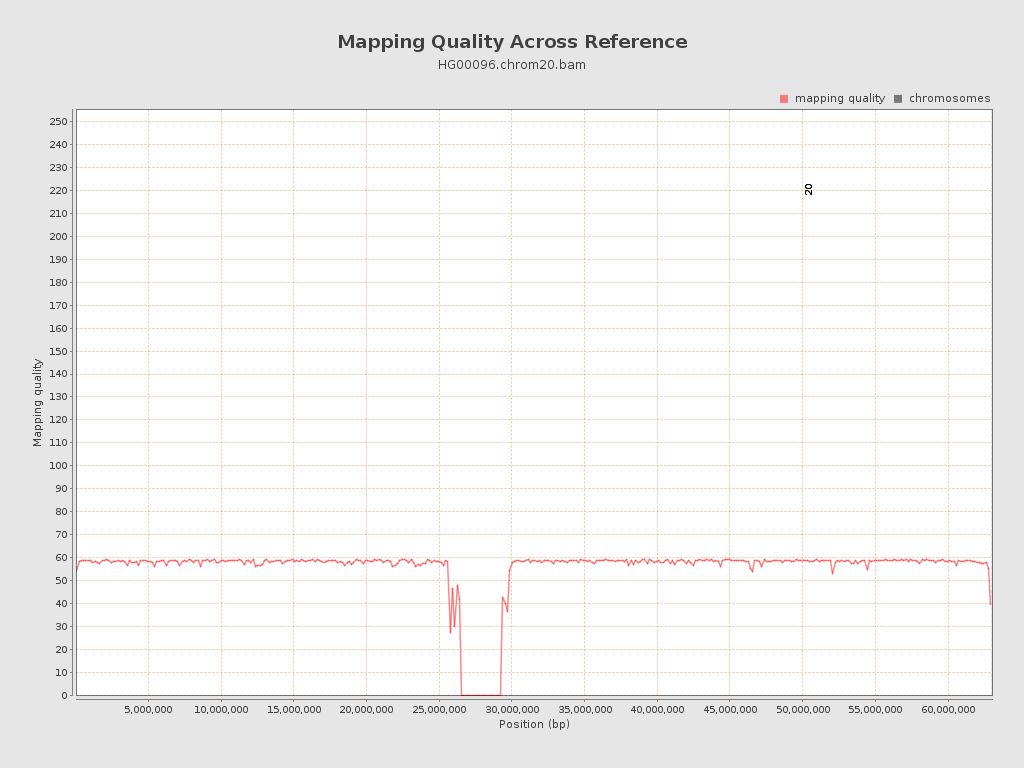
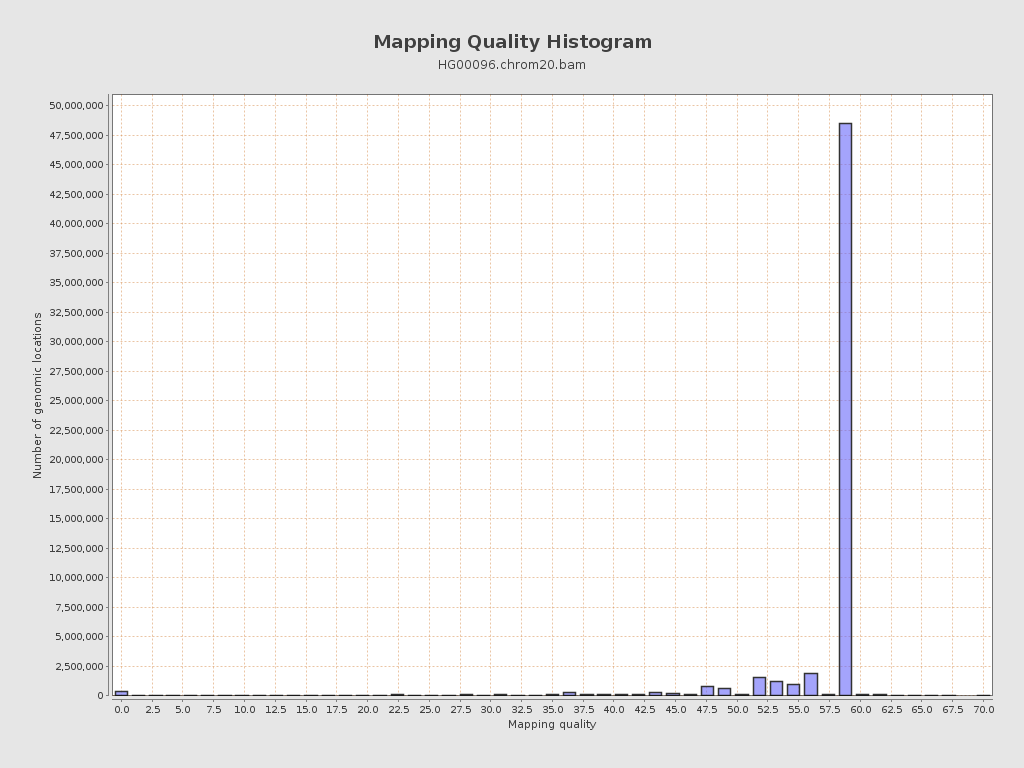
| Mean Mapping Quality | 55.35 |
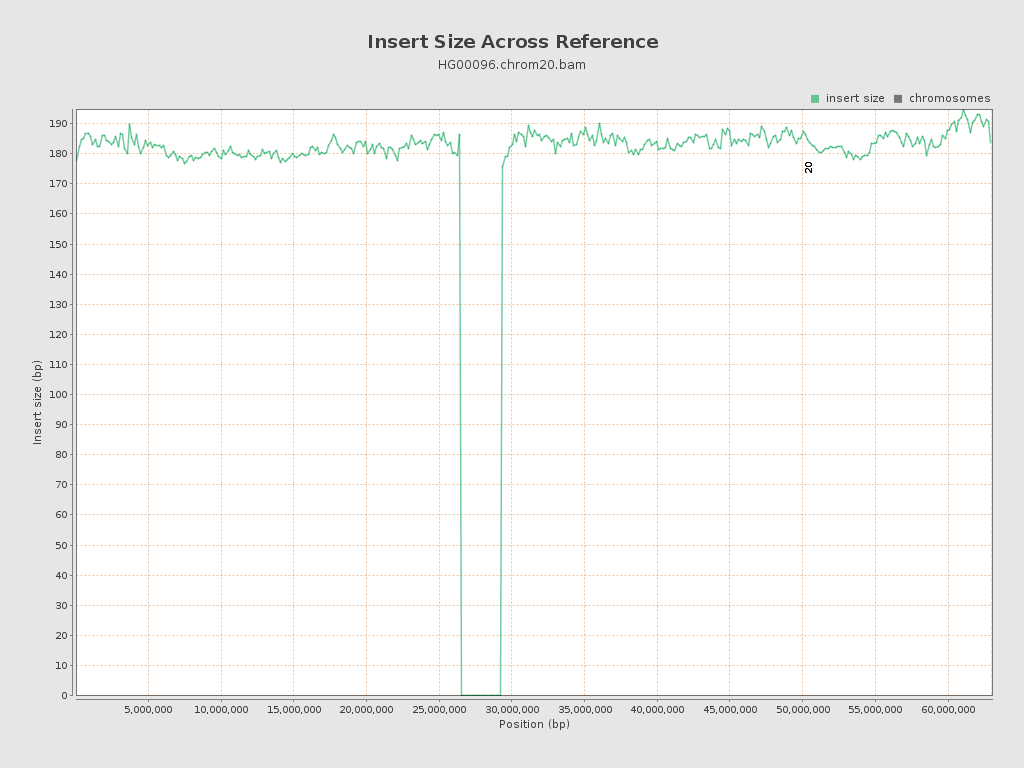
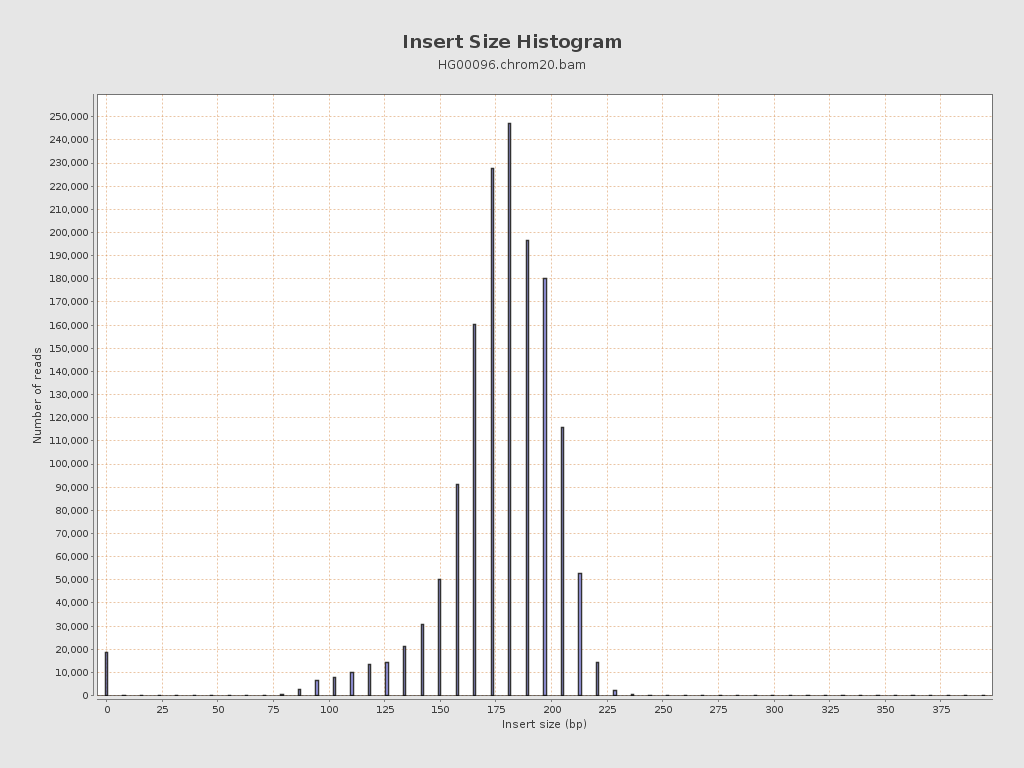
Insert size
| Mean | 3,128.51 |
| Standard Deviation | 295,452.44 |
| P25/Median/P75 | 171 / 184 / 197 |
Mismatches and indels
| General error rate | 0.35% |
| Mismatches | 938,327 |
| Insertions | 22,503 |
| Mapped reads with at least one insertion | 0.74% |
| Deletions | 27,083 |
| Mapped reads with at least one deletion | 0.89% |
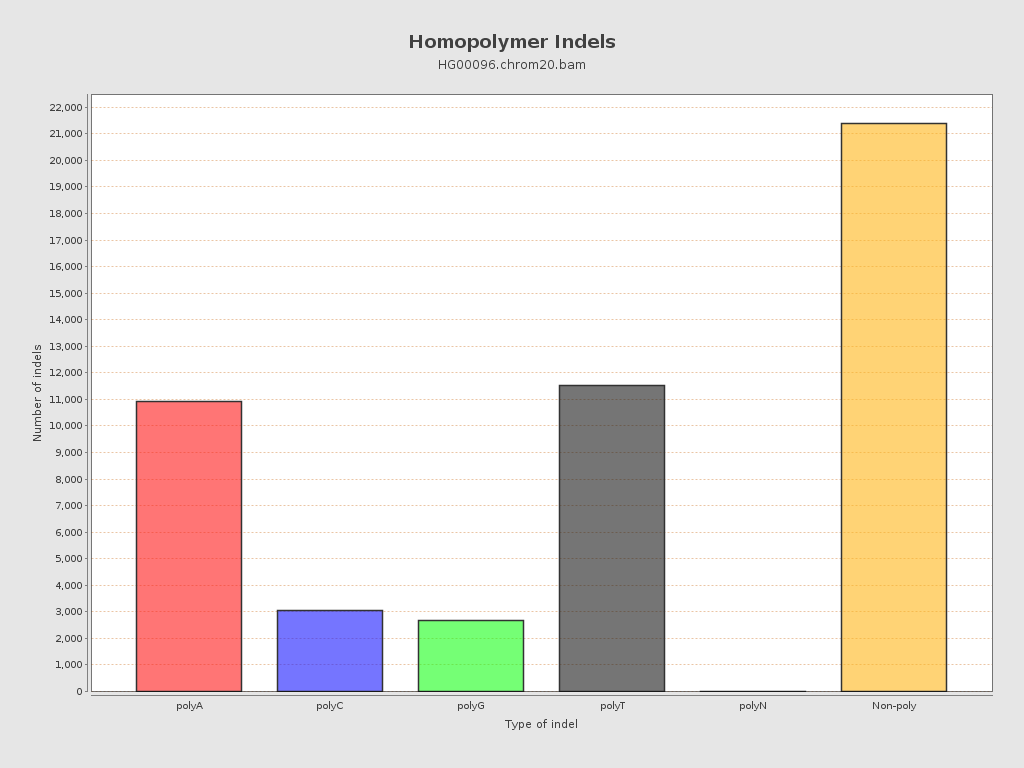
| Homopolymer indels | 56.85% |
Chromosome stats
| Name | Length | Mapped bases | Mean coverage | Standard deviation |
| 20 | 63025520 | 277945166 | 4.41 | 2.75 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}